The Helix Viral Sequencing Assay
Summary
- Helix has led scientific efforts to track and understand the evolution of SARS-CoV-2.
- This is a snapshot of Helix’s viral sequencing pipeline along with an assessment of its performance.
- Helix’s hybrid capture workflow is more robust to viral evolution than an amplicon approach.
Introduction
From the early detection of the Alpha variant in the United States, to the timely monitoring of the emergence of the Delta variant, Helix has been at the forefront of scientific efforts to track and understand the evolution of the SARS-CoV-2 virus. Our nationwide COVID-19 diagnostic testing footprint, enabled by partnerships with health systems, employers, and retail pharmacies, allows us to collect a geographically broad sample of SARS-CoV-2 positive samples. Since January 2021, Helix has partnered with the Centers of Disease Control and Prevention (CDC) to monitor the emergence of new SARS-CoV-2 variants in the United States. Helix has also partnered with Kaiser Permanente Southern California in their studies of real-world vaccine efficacy for the Pfizer and Moderna vaccines, providing high quality viral sequences and PANGO lineage and Nextclade clade designations for the COVID-19 positive cases in their cohort.
Helix began viral sequencing in-house in late May 2021. To date, we have sequenced over 120,000 samples and currently process thousands more per week. While we continue to improve and develop our assay, this white paper provides a snapshot of our current methods and details the due diligence and quality control measures put in place to ensure the integrity and quality of this high-throughput operation.
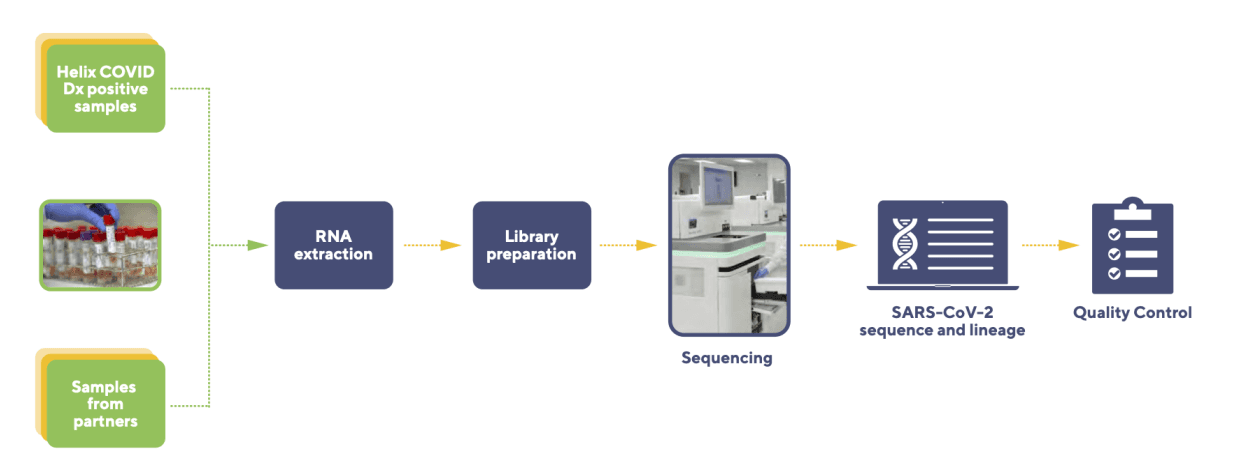
Figure 1: The Helix VSeq workflow.
VSeq workflow
Laboratory steps
We receive remnant specimens (Saline, Saliva, Universal Transport Medium) that tested positive for SARS-CoV-2 by qPCR from collaborators or from internal diagnostic testing at the Helix laboratory. Following accessioning, specimens are incubated in a laboratory oven using a preset protocol(s) that heat-inactivates the SARS-CoV-2 virus. Samples are then subject to RNA extraction, followed by RNA library preparation, and then SARS-CoV-2 genome capture. Next-generation sequencing is accomplished using the NovaSeq Sequencing system.
Bioinformatics Pipeline
The bioinformatics processing of the sequenced reads follows a standard series of steps for next-generation sequencing (NGS) data: demultiplexing with bcl2fastq (Illumina), aligning reads, calling variants, and generating a consensus sequence. We have made deliberate and data-driven choices in the details of each of these steps to ensure high-quality SARS-CoV-2 consensus sequences.
Following consensus sequence generation, a Pango lineage and clade designation is assigned using Pangolin (with the pangoLEARN model) and nextclade CLI, respectively. Updates to both tools are made on a regular basis, especially when a new variant of concern or interest is designated by the World Health Organization (WHO). All consensus sequences are then reprocessed on the newer version, ensuring that early occurrences of as-yet unnamed variants are later assigned the correct designation.
Quality Control
Quality control (QC) of the viral sequences occurs primarily at two levels: sample and plate. A sample-level QC status of ‘pass’ indicates a sample is unlikely to have been contaminated and has a sufficiently complete consensus sequence to be assigned a lineage. At the plate level, our QC criteria are designed to flag potential reagent issues or sample swaps that would require an entire plate to be re-processed (this is extremely rare).
Sequencing accuracy
During assay development, we confirmed the accuracy of the VSeq workflow with synthetic spike-in controls and simulated reads. Both types of controls led to consensus sequences with 100% identity to the expected strain. Synthetic controls are, however, of limited utility: there is a time lag between the discovery of a variant and availability of synthetic controls for that variant. Moreover, the conditions for success or failure of a sequenced synthetic control in R&D may not extrapolate to clinical samples in production. Our preference is therefore to evaluate our sequencing quality in real-time by comparing results from our clinical samples with national trends and with sequences generated by other nations/labs.
Trends in SARS-CoV-2 evolution in terms of PANGO lineage calls generated from Helix data have been extensively documented in both in our own publications (for Alpha and Delta variants), as well as in studies conducted with external investigators (for example, the Pfizer and Moderna vaccine efficacy studies). Here, we provide further data -- trajectories of key spike mutations -- so that the granularity of our sequencing assay is fully transparent (Figure 2). In particular, the S:G142D mutation, which is defining for the Delta variant, and which has been associated with primer artifacts in the ARTIC v3 design, correctly tracks in our data with the rise (and decline) of the Delta variant.
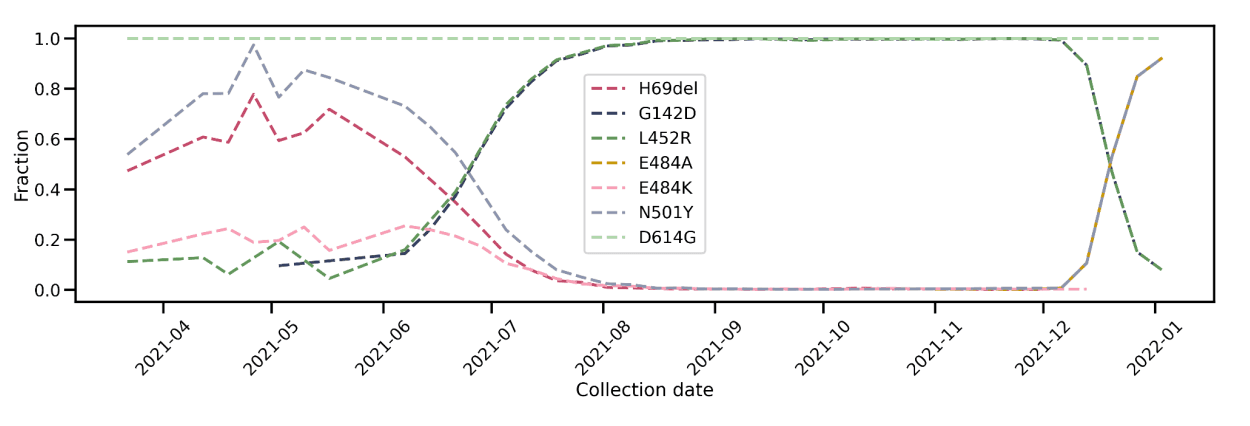
Figure 2: Prevalence of key Spike mutations over time. For the period 2021-12 to 2022-01, the trajectories of Omicron mutations H69del, E484A, and N501Y exactly overlap, as do those of Delta mutations G142D and L452R. (Sequence data prior to June 2021 generated by Illumina.)
Further assurance of the accuracy of our pipeline is the 100% prevalence of the S:D614G mutation in all of our data, consistent with its displacement of the S:D614 ancestral allele by the second half of 2020. We also see that the S:N501Y mutation, initially associated with the Alpha, Beta, and Gamma variants, along with S:H69del (Alpha and Eta/B.1.525), and S:E484K (Gamma and others) receded with the Delta wave from June to August 2021. With the recent Omicron surge, two of these mutations, S:N501Y and S:H69del, along with S:E484A, a new mutation unique to Omicron, have rapidly increased in prevalence. The S:L452R mutation, initially associated with the Epsilon variant (B.1.427 and B.1.429) and then a defining mutation for Delta, has followed the opposite pattern: it appeared in our data initially at low levels, consistent with the low nationwide prevalence of Epsilon, then increased to 100% of all sequences as part of the Delta wave, before declining with the recent Omicron surge.
Another external corroboration of sequencing accuracy is a phylogenetic analysis combining sequences originating from Helix’s diagnostic lab with sequences from other commercial or public health labs. Any systematic biases in Helix’s sequencing pipeline should manifest in a non-random distribution of Helix sequences in the phylogenetic tree. To this end, Figure 3 depicts the phylogenetic relationships for a random subset of sequences downloaded from GISAID, all originating from the state of Florida, USA, with collection dates in the week June 25-July 1, 2021. This period of time was chosen due to the high lineage diversity that week: Alpha (B.1.1.7), Delta (B.1.617.2), Mu (B.1.621), and Gamma (P.1) were all present at non-trivial frequencies. As can be seen, within each lineage, the samples originating from Helix are interspersed among samples originating from other labs, indicating that sequences generated by Helix’s workflow are just as similar to sequences generated by other labs as they are to other Helix-generated sequences.
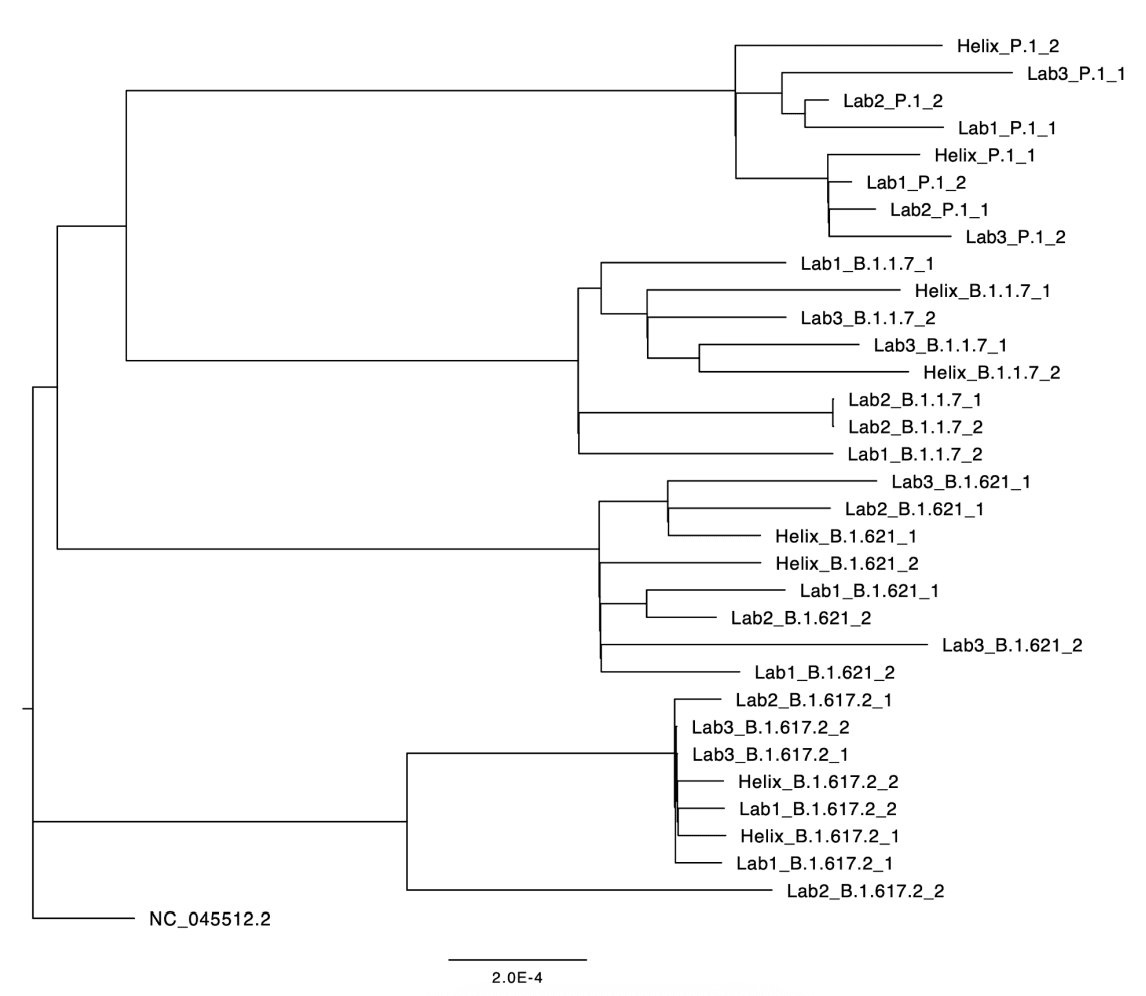
Figure 3: Phylogenetic tree of a random sample of sequences from GISAID all originating in Florida, June 25-July 1, 2021. Labs 1-3 are high-throughput commercial or public health laboratories (in no particular order: Lab Corporation, Quest Diagnostics, Florida Bureau of Public Health Laboratories). To minimize the chance that sequence similarity is confounded by geographic proximity, we randomly sampled two sequences for each lab/lineage combination (out of a median 27, range 9-82, sequences per lab/lineage combination).
Hybrid capture is preferable to an Amplicon workflow
We will close this white paper with a comparison of the performance of the hybrid capture and amplicon-based workflows. Whereas amplicon-based workflows appear to be the preferred protocol in SARS-CoV-2 sequencing, Helix has been intentional in using hybrid capture as the primary workflow, with the amplicon method as a supplement. We find that the advantage of the amplicon workflow lies primarily in its ability to generate higher coverage in samples with low viral titer (Figure 4a). But, when restricting to samples with medium-to-high viral titer, the hybrid capture approach is superior because it generates more complete sequences (Figure 4b) and is more robust to SARS-CoV-2 evolution (Figure 4c). This advantage has become even more evident with the rapid rise of the highly mutated Omicron variant.
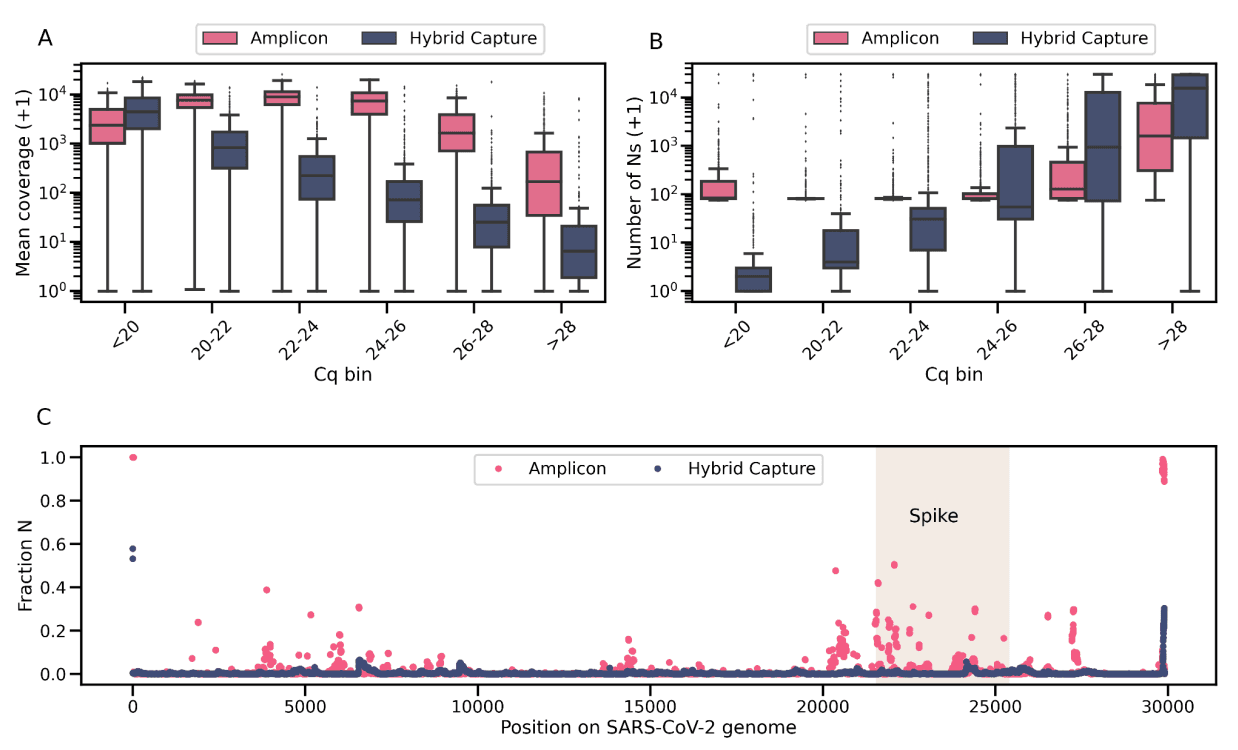
Figure 4: Comparison of the two workflows. a) Mean SARS-CoV-2 genomic coverage across Cq bins for all samples. b) Number of uncertain bases (N) across Cq bins for all samples. For both a) and b), because of the log scale, the y axis plots the variable + 1. c) Fraction of passing sequences with an N by position in the SARS-CoV-2 genome. The shaded rectangle corresponds to the region that encodes the S glycoprotein.
As Figure 4b illustrates, even at low Cq values, the amplicon workflow generates sequences that have more Ns than the hybrid capture workflow. First, there is a minimum of 75 uncertain bases (Ns) due to 25 bp on the 5’ of the genome and 50 bp on the 3’ end of the genome which are not covered by the non-primer portion of any amplicon. Second, with the amplicon method, many positions within the SARS-CoV-2 genome drop out, i.e. there are intermittent regions of zero read coverage: multiple positions in the Spike region have >20% of passing consensus sequences reporting an N at that position (Figure 4c). In contrast, the hybrid capture workflow exhibits much lower levels of drop-out, and the completeness of the consensus sequence decreases in a predictable manner with increased Cq values, because the increase in Ns is primarily due to lower genome-wide coverage. For these reasons, and especially due to the highly mutated and dominant Omicron variant, Helix will run samples exclusively through the hybrid capture workflow going forward.
Authors
Shishi Luo, Andrew Dei Rossi, Efren Sandoval
Categories